
Home
ꄲ
Alioth Knowledge Center
ꄲ
2025 NMPA Annex on Sterile Medicinal Products: Requirements for Sterilizing Filtration and Challenges Faced by Enterprises
2025 NMPA Annex on Sterile Medicinal Products: Requirements for Sterilizing Filtration and Challenges Faced by Enterprises
法规背景
2025年3月17日,国家药监局(NMPA)正式发布《无菌药品附录(征求意见稿)》,这是继2011年首版无菌药品生产规范发布14年以来的首次系统性修订。此次调整背后,是中国药品监管深度融入国际体系的战略布局:2023年NMPA以高标准通过PIC/S(国际药品检查合作计划)正式申请者资质审核,而此次修订内容更是充分对标国际——不仅与2022年更新的《欧盟GMP附录1:无菌药品的生产》技术要求全面接轨,更呼应了PIC/S成员国对无菌药品生产的前沿监管要求。

新版附录以"设计合规"为核心逻辑,系统梳理了厂房设施、工艺流程、灭菌方法等11大模块的升级要求。尤其在风险控制维度,新增对无菌生产全链条的精细化管理指标,其中除菌过滤(Sterilizing Filtration)部分完整吸纳欧盟对除菌过滤工艺的验证体系(涵盖完整性测试、冗余过滤等),填补了国内技术规范空白。这一变革既体现了我国制药行业向国际最佳实践看齐的决心,也为NMPA最终加入PIC/S奠定了技术标准基础。

 表1.无菌药品附录更新对比
表1.无菌药品附录更新对比
法规升级看点:2025新规 VS 2011版的差异
系统设计更科学
- 新增:过滤器材质必须与药液相容性验证(第179、181条)


- 收紧:除菌过滤系统布局需结合微生物负荷监测(第180条)


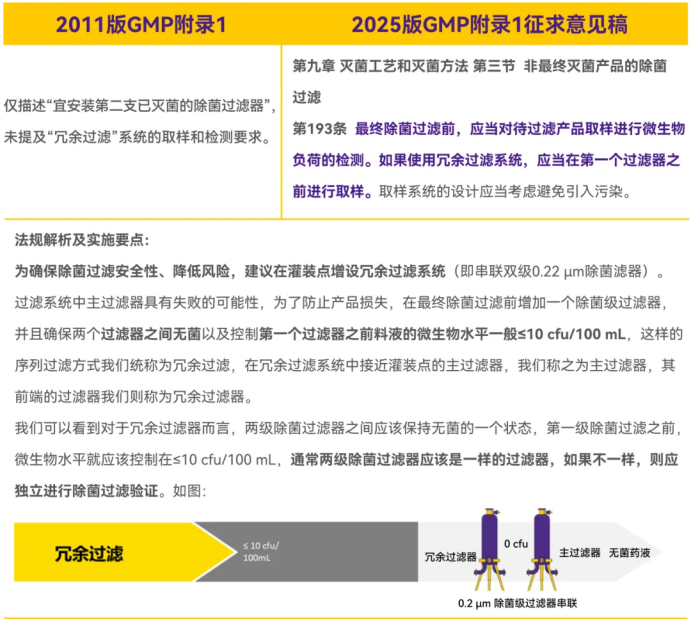
- 微生物监控:冗余系统需在首个过滤器前进行微生物负荷取样(第193条)

- 升级:验证过滤参数需实时记录并调查偏差(第185、186条)


完整性测试全链条管控
- 关键环节:过滤器使用前灭菌后完整性测试PUPSIT(第187条)


- 例外管理:微量药液场景允许风险评估替代方案(第187条补充)


- 双保险策略:不同序列过滤方式下过滤器完整性检测要求(第191、192条)


- 气体过滤:关键无菌气体(如氮气)过滤器同标准管理(第188条)


使用时限精细化管理
- 时段监控:最长单次使用限1天,多批次生产需严格审批(第194条)


- 验证强制:每款产品需独立验证过滤器最长使用时间(第195条)


风险控制再升级
- 整合至CCS:所有过滤管理必须纳入企业污染控制策略(第180条)


- 场景覆盖:冻干机密封性过滤器需灭菌+测试双重保障(第128条)


总结
2025版NMPA无菌附录强化除菌过滤要求:建议实施冗余过滤系统,推行PUPSIT测试,构建实时参数监测与兼容性-微生物负荷控制-过滤时限等维度验证体系,并深度整合至CCS框架。新规强化A级区动态管控,要求企业重构工艺验证平台并建立供应链质量联动机制。作为专业过滤技术合作伙伴,我们提供过滤器全生命周期验证服务及战略级合规咨询,赋能企业精准达成新规要求。
