
A Discussion on Bacteria Reduction Filtration Based on the New Edition of the "Good Manufacturing Practice (GMP) Guide for Pharmaceutical Products"
近年来,药企越来越重视质量风险管理,加之法规、指南和标准的完善,以及监管的强化,使得除菌过滤在无菌制剂生产中的关键作用得到了广泛认可。然而,除菌过滤前有一个重要环节很容易被忽视:减菌过滤,它是除菌过滤前的一个关键步骤。
除菌过滤器广泛应用于无菌药品的生产过程中,根据过滤的目的不同,将过滤分为除菌过滤和减菌过滤。
什么是减菌过滤?
不同于除菌过滤,减菌过滤旨在通过物理截留的方式,将待过滤介质中的微生物污染水平降低至一个可接受的程度。
“对无菌药品生产的全过程进行微生物控制,避免微生物污染。最终除菌过滤前,待过滤介质的微生物污染水平一般小于等于10CFU/100mL。”---NMPA(国家药品监督管理局)2018年第85号《除菌过滤技术及应用指南》
减菌过滤通常应用于两个关键节点
一是在终端灭菌工艺生产的无菌制剂灌装前,二是在非最终灭菌工艺生产的无菌制剂的除菌过滤工序前。其核心目的在于确保产品在最终灭菌或除菌过滤前,微生物污染水平达到预期标准,以保障过滤过程的清洁度和安全性。

除菌过滤与减菌过滤
除菌过滤和减菌过滤在过滤系统中各有分工,但也共享一些控制要求。
共同点包括:
- 两者都要限制微粒和内毒素的释放,防止液体再污染
- 均需控制滤芯材料可能释放的可提取物,以免影响产品质量
不同之处在于:
- 除菌过滤旨在彻底清除微生物,保持产品无菌,但孔径小,易堵塞
- 减菌过滤则旨在减少而非清除微生物,孔径较大,减少堵塞风险
除菌过滤和减菌过滤的特性比较
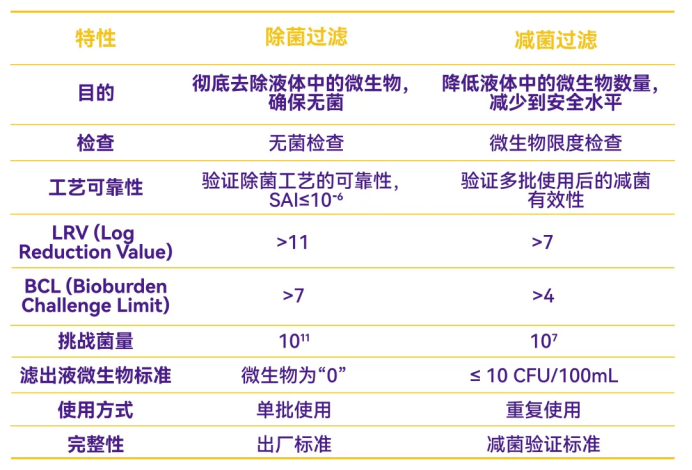

综上,在实际应用中,这两种过滤工艺往往是结合使用的。例如,在无菌制剂的生产中,可能会先使用减菌过滤器预处理药液,以降低微生物负荷,然后再通过除菌过滤器确保最终产品的无菌性。
减菌过滤工艺
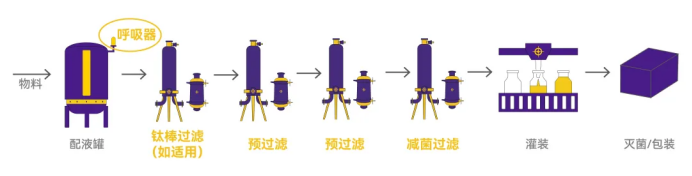
在最终灭菌工艺中(见Figure 1),物料首先在配液罐中被处理,然后通过钛棒过滤(如适用)进行初步过滤。药液经过两级预过滤以进一步清除较大颗粒,之后通过减菌过滤器降低微生物数量。最后经过灌装和灭菌/包装步骤,确保产品的无菌性。
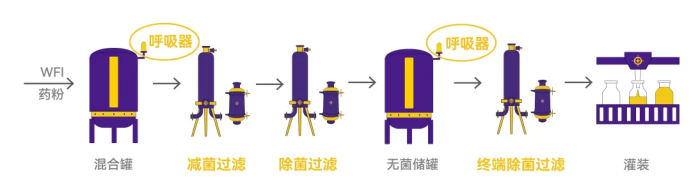
在非最终灭菌工艺中(见Figure 2),WFI(注射用水)和药粉在混合罐中混合,然后通过减菌过滤器去除微生物。药液通过除菌过滤器进一步确保无菌,再存储在无菌储罐中。在灌装前进行终端除菌过滤,以确保最终产品的无菌性。
 点击查看大图
点击查看大图
Figure 1. 减菌过滤器在最终灭菌工艺中的应用

点击查看大图
Figure 2. 减菌过滤器在非最终灭菌工艺中的应用
总结来说,减菌过滤在两种工艺中都起到了至关重要的作用,它不仅减少了微生物的负荷,还为后续的除菌过滤提供了保护,确保了药品的无菌性和安全性。通过合理的过滤流程设计,可以提高过滤效率,降低生产成本,并确保药品质量。
减菌过滤系统设计
减菌过滤一般被设计在灌装或除菌过滤之前。其主要目的是减少药液中的微生物数量至一个可接受水平,而不是完全去除微生物。但减菌过滤可有效控制微生物污染,并且能够去除杂质颗粒。减菌过滤的使用还有助于在湿热灭菌前降低微生物污染,从而减少灭菌后的热原水平。
过滤工艺及系统设计-冗余过滤
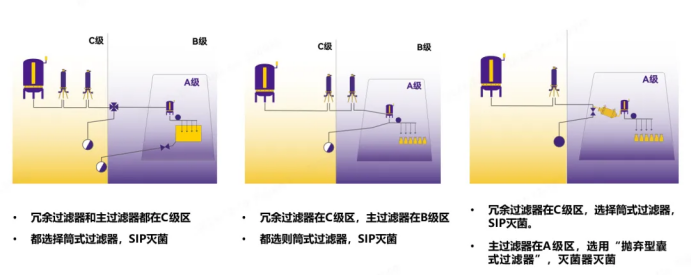
液体过滤器以下三种形式较为常见,但设计方式不止如此(Figure 3)。

点击查看大图
Figure 3. 冗余过滤系统三种常见设计
*气体过滤器根据产品工艺需求,一般安装在洁净环境A、B、C、D级区,如需要,也可以安装在一般区,如灭菌柜的压缩空气过滤器。
过滤器的硬件配置
当条件允许时,全封闭的并能在线灭菌和完整性测试的系统对无菌工艺方面是第一选择。
过滤系统一般组成包括: 过滤器前药液罐、过滤后药液罐、泵、完整性测试仪、过滤器(套筒、过滤芯)、压力测定装置(压力表或压力传感器)、温度记录装置(温度表或温度传感器)、过滤管线、冷凝水处理装置(如疏水阀)等。
无菌产品过滤所用的不锈钢套筒的一般要求为:
- 接触药液的部分应选用316L不锈钢材质
- 耐高压和高温,例如:121°C,耐压150psi(约10bar)
- 卫生级夹具的进口和出口
- 卫生级软管接口的排气和排水
- 易于冷凝水的排放
- 易于冷空气的排放
- 药液残余少
- 磨制/电解质抛光
- 兼容性较好的垫片和O型圈

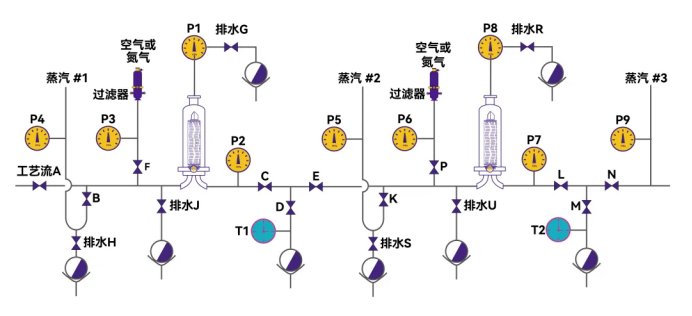
点击查看大图
Figure 4. 某冗余过滤系统的阀门管道设计
过滤后储罐位置的要求:对于过滤系统中,过滤后药液罐或缓冲罐的位置有多种形式,具体见Figure 5。

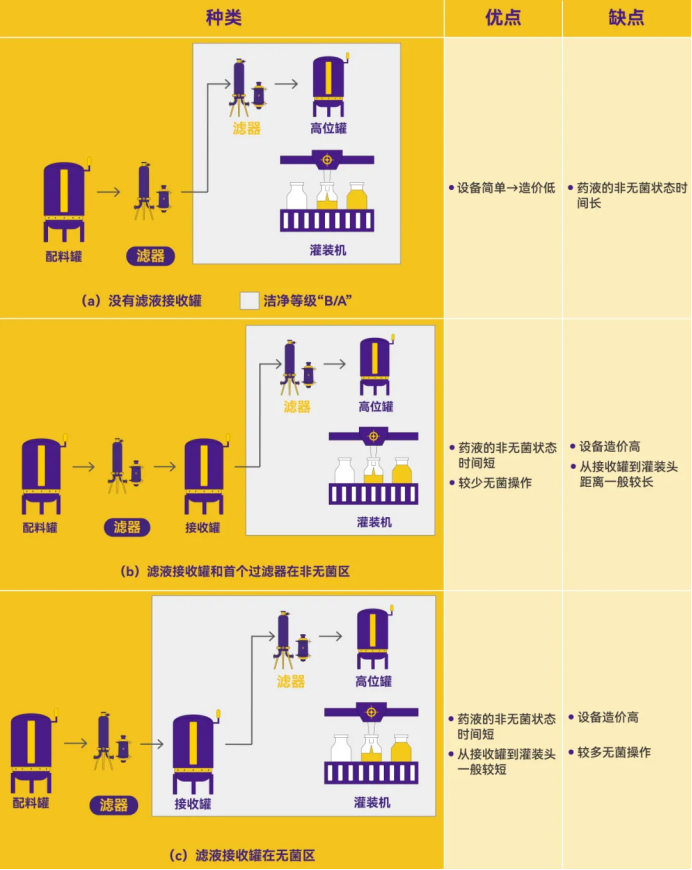
点击查看大图
Figure 5. 过滤器与储罐的三种设计及优缺点
减菌过滤系统应使用0.45μm或0.22μm(或以下)的过滤器来降低微生物污染。设计时要考虑过滤器大小、药液量、过滤时间和压差等因素,确保过程可控。同时,要监控药液在过滤过程中的微生物水平,因为药液在过滤前后不是无菌的。
减菌过滤器的重复使用
液体减菌过滤器设计用于单次或连续多批次生产。在实际操作中,它们有时用于同一产品的多次生产。重复使用通常指同一过滤器用于同一液体产品的多个批次。以下情况被视为重复使用:
- 批次间进行冲洗
- 批次间冲洗和灭菌
- 批次间清洗、保存和灭菌
在决定是否重复使用过滤器时,应基于对产品和工艺风险的深入了解,并通过风险评估来做出判断。考虑的风险因素包括:
- 细菌的穿透
- 过滤器完整性缺陷
- 可提取物的增加
- 清洗方法对产品内各组分清洗的适用性
- 产品存在的残留(或组分经灭菌后的衍生物)对下一批次产品质量风险的影响
- 过滤器过早堵塞
- 过滤器组件老化引起的性能改变等
过滤器的重复使用需经过验证,考虑药液特性(如pH、浓度)、过滤和灭菌条件、使用次数及批量。选择最严苛条件进行测试,以确保过滤器的安全性和有效性。过滤器重复验证项目见Figure 6。


点击查看大图
Figure 6. 过滤器重复使用验证项目
综上,针对采用减菌过滤工艺,对灭菌前的微生物污染水平应控制在可接受范围内,过滤器重复使用不应降低微生物污染控制水平。
过滤器重复使用应在验证工艺参数范围内,在使用过程中应监测消毒/灭菌次数、过滤批量、压力/上下游压差、温度、过滤器使用次数以及完整性测试结果等。
减菌过滤系统过滤器验证
在进行过滤工艺验证时,需要考虑滤芯材质与产品之间的化学兼容性,以及滤芯可能迁移出的物质对终端制剂的影响。同时,根据NMPA《除菌过滤技术及应用指南》和EU GMP Annex-1的要求,验证过程应包括化学兼容性、可提取物/浸出物及吸附等测试。
“减菌过滤的正常运行是保证产品最终灭菌前(或除菌过滤前)的微生物污染水平符合可接受程度的重要措施。减菌过滤工艺验证应包括化学兼容性,可提取物/浸出物及吸附等。”—2018年NMPA第85号《除菌过滤技术及应用指南》
“8.81 The selection of components for the filtration system and their interconnection and arrangement within the filtration system, including pre-filters, should be based on the critical quality attributes of the product, justified and documented.”过滤系统组件的选择及其在过滤系统内的相互连接和布置,包括预滤器,应基于产品的关键质量属性,并经过证明和记录。--2022年EU GMP Annex-1
综上所述,无论是为了管理工艺风险还是满足法规要求,减菌过滤器的验证都是一个需要考虑的环节。
详细验证部分可关注后续分享内容“液体除菌过滤的确认和验证”,包括除菌过滤器的确认、过滤工艺验证、确认与验证建议、验证项目等。
结论
减菌过滤是无菌药品生产中的一项关键技术,它通过精确控制微生物污染水平,为药品的质量和患者的安全提供了有力保障。随着制药技术的发展,减菌过滤将继续在无菌药品生产中发挥其重要作用。
参考法规与指南
[1] 《药品GMP指南》(2023年修订)无菌制剂-上册;
[2] PDA Technical Report No.26 ,Revised 2008,Sterilizing Filtration of Liquids;
[3] EU Guidelines to Good Manufacturing Practice,Annex 1 Manufacture of Sterile Medicinal Products,2022.
