
Practical Guidelines for Sterilizing Filtration with Disinfectants
根据GMP附录1《无菌药品》规范,药品生产的洁净区实行梯度管理标准,其中A级和B级洁净区作为核心无菌操作区域,需实施动态微生物监测。第四十四条要求应当监测消毒剂和清洁剂的微生物污染状况,配制后的消毒剂和清洁剂应当存放在清洁容器内,存放期不得超过规定时限。A/B级洁净区应当使用无菌的或经无菌处理的消毒剂和清洁剂。


近期FDA警告信显示,某企业无菌注射剂产品在ISO 5级区使用非无菌消毒剂【Your firm used a non-sterile disinfectant within the ISO 5 aseptic processing area:在ISO 5级无菌操作区域内使用了非无菌消毒剂】,此违规行为不仅直接违反GMP核心要求,更暴露出洁净区管理中存在基础控制失效风险。这类关键区域的管控疏漏将直接影响药品无菌保障水平,危及患者用药安全。
因此,在无菌生产核心区域引入非无菌消毒剂的操作,实质上构成了对受控环境微生物管控体系的直接挑战。
消毒剂为什么要进行除菌过滤?
医疗领域消毒剂的效力等级划分(高效、中效、低效)本质上揭示了其杀菌谱差异——由于消毒性能存在选择性,所以该分类体系意味着任何消毒制剂都无法实现全微生物谱系的灭活能力。需特别指出的是,“消毒剂”作为集合概念包含多个功能性亚类,欧盟法规对此进行了严格的功能分类:根据目标微生物类型细分为杀菌剂(Bactericides)、杀真菌剂(Fungicides)、杀孢子剂(Sporicides)等法定类别。

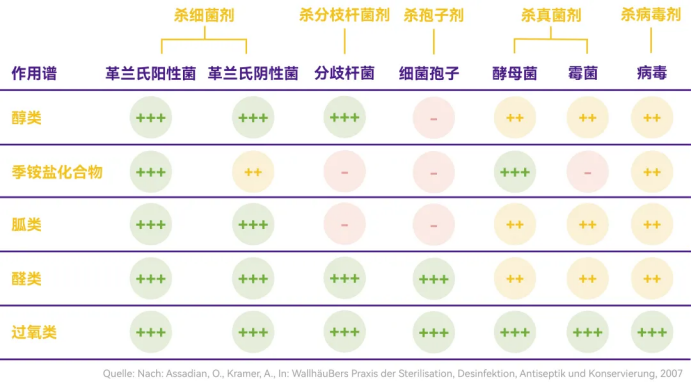
图示:消毒剂覆盖微生物谱类型
由此可见,消毒剂未杀灭的微生物将残存,部分菌种可形成生物膜。对于洁净级别高的A、B级区,甚至有些C、D级都需考虑使用无菌消毒剂,以确保洁净区无菌状态。
消毒剂与除菌滤器之间的适用性评估
根据《除菌过滤技术及应用指南》,进入A/B级洁净区的消毒剂可以采用除菌过滤的方法除菌,但应评估消毒剂与所选择滤器材质之间的适用性,滤器使用后需进行完整性测试。
消毒剂与过滤器之间的化学兼容性应作为过滤器选择时的关键考量点之一。
艾里奥斯除菌滤器与消毒剂兼容性验证案例

实验设计与方法
1. 测试对象
· 滤器规格:Alipore® DHC双层PES膜,孔径0.45/0.2μm
· 消毒剂类型:75%乙醇/95%乙醇、70%异丙醇/复合醇、3%杀孢子剂、6%过氧化氢/复方过氧化氢、复方季铵盐(H)、0.1%新洁尔灭(苯扎溴铵)、3%来苏尔(甲酚和钾肥皂)
2. 实验流程
① 预处理:121℃高压蒸汽灭菌30分钟→纯水润湿→检测初始完整性(起泡点≥3520mbar)与流速初始值
② 浸泡处理:室温下连续接触消毒剂7天(模拟极端工况)
③ 后处理检测:
· 物理表征:目视检查(色变/形变)+ 扫描电镜(SEM)膜结构分析
· 性能测试:完整性、流速变化率、重量损失率
关键实验结果
1. 完整性验证
所有测试组均通过扩散流与泡点双重标准,其中典型数据包括:
· 极端溶剂组:95%乙醇接触后起泡点4750mbar(初始值4667mbar)
· 强氧化剂组:6%过氧化氢扩散流13.4mL/min(初始值14.1mL/min)
2. 其他测试参考-稳定性评估

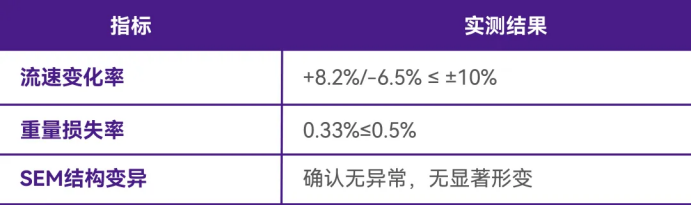
结论与应用建议
- 兼容性确认
艾里奥斯Alipore® DHC PES过滤器与本次测试的几大类消毒剂在室温条件下接触7天后,完整性测试通过,外观无颜色变化、形变、破损、裂纹等现象,结合过滤器接触料液前后的重量变化、水流速变化及滤膜表面的扫描电镜数据,可以说明艾里奥斯生产的PES过滤器与上述消毒剂有良好的化学兼容性。
- 验证扩展
以上测试结果是对艾里奥斯的Alipore® DHC PES过滤器在消毒剂除菌过滤适用性的有力支持。但在实际应用中企业需综合评估消毒剂的应用场景、具体过滤工艺以及料液特性等(温度、接触时间、消毒剂批次差异),判断是否需要进行更加深入的评估或研究,以降低无菌产品生产风险,满足法规要求。
选择合适的消毒剂滤芯
鉴于消毒剂本身具有化学氧化与腐蚀特性,其与滤芯材料存在不兼容风险。根据GMP过滤系统验证指南要求,必须针对所使用材质(如PES、PTFE)开展化学兼容性测试,并通过验证报告证明其长期接触消毒剂时,物理完整性及过滤效能均符合工艺限度标准。
常用消毒剂除菌滤器推荐


关于推荐表中适配消毒剂的滤芯品类,艾里奥斯已同步验证其兼容性和完整性测试,具体适用性测试(温度、接触时间、压力等),建议制药企业在选型阶段与供应商开展技术规格对标,同时结合消毒剂类型及生产工艺需求以评估风险,构建符合法规要求的消毒剂过滤体系质量文件。
参考法规与指南
[1] 国家食品药品监督管理局. 药品生产质量管理规范(2010年修订). 2011.
[2] 国家食品药品监督管理局. 药品生产质量管理规范(2010年修订). 无菌药品附录.2011.
[3] 国家药品监督管理局. 除菌过滤技术及应用指南(2018年第85号通告). 2018-07-31.
[4] FDA (2004) Guidance for Industry, Sterile Drug Products Produced By Aseptic Processing – Current Good Manufacturing Practice.
[5] EU Guidelines to Good Manufacturing Practice. Medicinal Products for Human and Veterinary Use, Annex 13. Investigational Medicinal Products, February 2010.
