
Safety Assessment: Key Points Introduction of Extractables & Leachables
前几期“滤”见新知系列我们介绍了关于除菌滤器的验证概念及验证主计划和细菌截留测试,本篇主要介绍核心验证项目之一:可提取物&浸出物以及安全性评估。
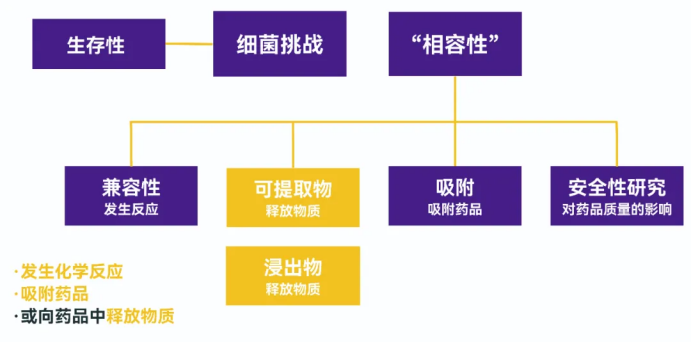
本文分为5个部分:
- 何谓可提取物?浸出物区别在哪?
- 杂质来源有哪些?受何因素影响?
- 如何开展可提取物/浸出物验证?
- 验证结果如何分析?
- 安全性评估怎么做?
何谓可提取物?浸出物区别在哪?
在NMPA的《除菌过滤技术及应用指南/2018》中有以下定义:
可提取物 -(Extractable)
在极端条件下(例如有机溶剂、极端高温、离子强度、pH、接触时间等),可以从过滤器及其它组件材料的工艺介质接触表面提取出的化学物质。可提取物能够表征大部分(但并非全部)在工艺介质中可能的潜在浸出物。
浸出物 -(Leachable)
在存储或常规工艺条件下,从接触产品或非接触产品的材料中迁移进入药物产品或工艺流体中的化学物质。浸出物可能是可提取物的一个子集,也可能包括可提取物的反应或降解后产物。
杂质来源有哪些?受何因素影响?
直接来源:滤器材质本身
滤膜:基础材质(如PVDF、PES)及添加剂(增塑剂、抗氧剂、表面活性剂等)
其他组件:端盖、滤壳、密封圈等非滤膜部件
间接来源:工艺条件影响
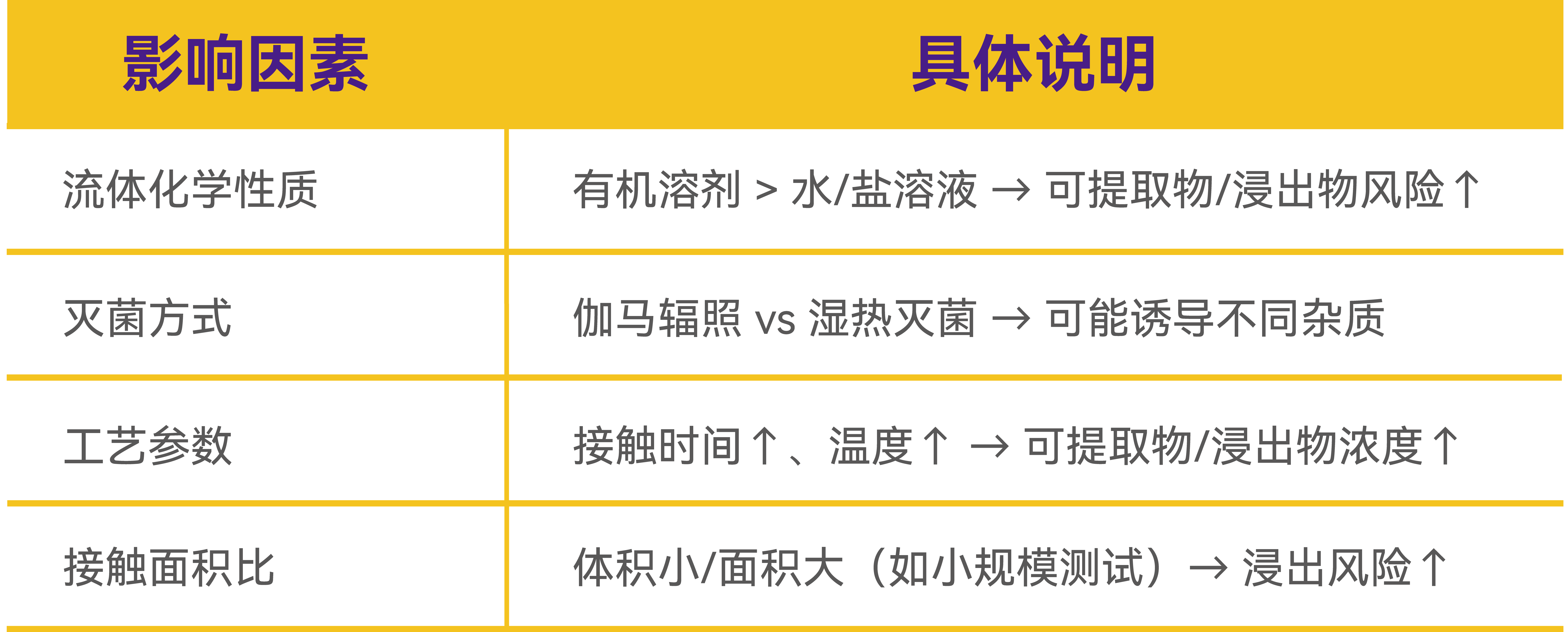

关键提醒:验证需覆盖整个过滤系统以及结合工艺条件考量,不局限于单一滤膜!
如何开展可提取物/浸出物验证?
验证之前先进行风险评估
“浸出物存在于最终原料药和药品中,通常包含在可提取物内,但由于分离和检测方法的限制以及浸出物的量极小,很难被定量或定性。应先获得最差条件下的可提取物数据,将其用于药品的安全性评估。可提取物反映了浸出物的最大可能,无论是否要做浸出物试验,可提取物的测试和评估都非常重要。”-- NMPA《除菌过滤技术及应用指南/2018》
从监管评估视角,可提取物数据不可缺少。 虽然浸出物直接反映实际应用场景(含背景干扰),且随着检测能力提升更具可操作性,CED指南特别强调:可提取物代表浸出物的理论最大暴露风险(最差条件)。二者实为互补而非替代——可提取物划出安全阈值边界,浸出物确认日常水平,共同构成风险评估体系。
可提取物的风险评估考量
可提取物实验前需做风险评估,非所有过滤工艺都需复杂验证,关键评估维度如下表:


可提取物的风险评估-关键评估维度
基于风险评估的可提取物验证试验
根据USP 665建议,不同风险等级的验证实验主要分为两类:
1. 生物反应性测试
低风险豁免|中风险测细胞毒性|高风险增加体内测试
2. 可提取物实验
·模型溶剂选择:低风险与中风险样品均使用50%乙醇水溶液;高风险样品需额外增加pH=3的酸溶液和pH=10的碱溶液。
·分析内容分层级:
A.低风险&中风险:基础测试包括NVR(非挥发性残留物)和UV吸收。
B.中风险:在基础测试上增加有机提取物测试。
C.高风险:需结合多种溶剂(50%乙醇、酸、碱溶液)提取物,进行综合测试。
·分析方法扩展:除低/中风险的分析项目外,高风险测试中必要时需补充元素测试。
需要注意的是,上述方案主要适用于常规工艺场景,无法覆盖所有情况。例如:
-有机相浓度>50%:此时采用50%乙醇水溶液进行提取可能不再适用;
-极端pH(>10 或 <3)溶液。
针对此类情况,我们需根据实际工艺条件,定制化设计实验方案,确保可提取物研究更精准、更全面地满足法规要求。
验证结果如何分析?
可提取物和浸出物的检测方法包括定量和定性两类,如 LC/PDA/MS, DI-GC/MS, HS-GC/MS, ICP/MS, UV, FTIR, NVR, TOC,为了保证分析方法的可靠性,需对分析方法进行验证或确认。选择哪几种分析方法,取决于实际的药品和生产工艺以及过滤器生产商对过滤器的充分研究。
具体分析方法详见下表:

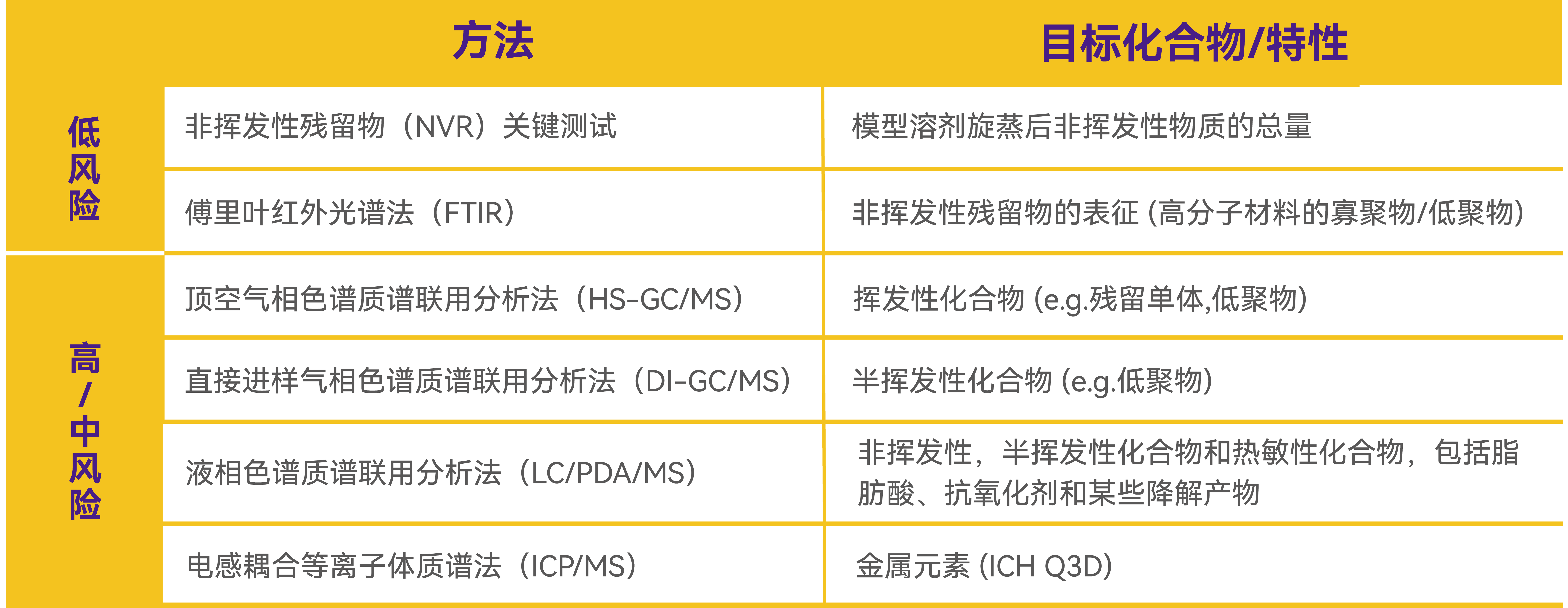
基础 & 完整可提取物分析方法
浸出物试验研究
浸出物实验通常在可提取物的安全性评估显示其对患者风险不可接受时启动。
进行浸出物研究前,需执行系统适用性研究,此步骤比较重要,用以确认分析方法的可行性,克服在实际药品中可能存在的干扰物影响。
其次,是浸出物的分析:
1.基于可提取物数据预测潜在目标浸出物
2.优先关注:
·可提取物研究中检出量高的物质
·检测灵敏度较低的物质(避免漏检)
·高毒性物质(即使含量低也可能风险显著)
安全性评估怎么做?
为什么要开展安全性评估
- 基于安全方面的考虑和监管部门的要求
“在完成可提取物或浸出物试验后,应针对过滤器可提取物或浸出物的种类和含量,结合药品最终剂型中的浓度、剂量大小、给药时间、给药途径等对结果进行安全性评估,以评估可提取物和浸出物是否存在安全性风险。” --《除菌过滤技术及应用指南》,NMPA, 2018年10月
安全性评估流程
完成可提取物和浸出物研究后,需对检出的杂质进行系统性安全性评估。其核心步骤如下:
1. 杂质分析与暴露量计算
杂质分析:通过可提取物/浸出物研究明确杂质的种类和定量水平。
暴露量计算:结合药品的临床使用参数(如剂量、剂型、给药周期),计算患者对杂质的每日暴露总量(Total Daily Exposure)。
2. 毒理学评估
评估执行:由具备资质的毒理学家根据暴露量数据开展安全性评估。
评估方法:
A.毒理学关注阈值(TTC, Threshold of Toxicological Concern):适用于缺乏特定毒理学数据的化合物。
B.允许每日暴露量(PDE, Permitted Daily Exposure):适用于具备充分毒理学数据的化合物。
方法选择:TTC或PDE的适用性需根据物质特性与数据完整性科学判定。
3. 风险判定与风险控制
可接受风险:若暴露量 < TTC或PDE值 → 风险可忽略,过滤器可安全用于生产。
不可接受风险:若暴露量 > TTC或PDE值 → 需启动风险控制措施:
A.工艺优化:增加稀释因子、调整工艺参数降低杂质水平;
B.分析升级:采用高灵敏度方法(如GC-MS/LC-MS)重新检测;
C.再评估:控制措施实施后,重新执行安全性评估。

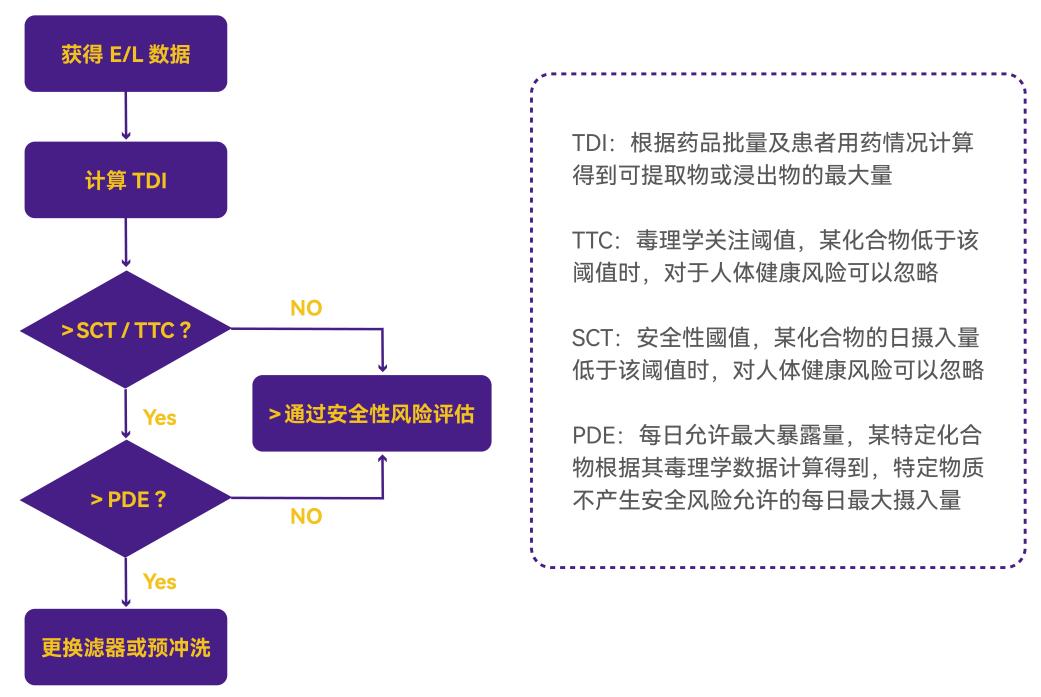
基于过程的安全性评估策略总结
1. 风险初评
根据过滤器配置、工艺条件等设计可提取物研究方案。
2. 安全决策树
·风险可忽略(可提取物数据支持):直接提交监管申报。
·风险不可忽略:执行浸出物定量分析;或实施风险控制措施后重新评估。
3. 监管提交
最终确认风险可控,提交完整证据链至监管机构。
总结
本期要点聚焦过滤系统安全性评估的核心——可提取物与浸出物,可提取物(最差条件下提取的化学物质)和浸出物(实际工艺下迁移的杂质)是评估过滤系统安全性的关键,需基于风险等级(溶液性质、工艺条件、制品风险)选择模型溶剂(基础:50%乙醇;高风险+酸碱液)和分析内容(基础:NVR+UV;升高:有机物+元素)。最终通过计算患者每日暴露量,比照TTC/PDE毒理学标准进行安全性判定,确保药用安全。
参考法规与指南
[1]《药品生产质量管理规范》(2010年修订)。
[2]《除菌过滤技术及应用指南》(国家药品监督管理局2018年第85号)。
[3] Bio-Process System Alliance (BPSA) Recommendations for Testing and Evaluation of Extractables from Single-Use Process Equipment, 2010.
[4] USP PF <665>, Plastic Materials, Components, and Systems Used in the Manufacturing of Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Product.
[5] BPOG: Best Practices Guide For Extractables Testing of Polymeric Single-Use Components Used in Biopharmaceutical Manufacturing, 2020.
[6] ICH HARMONISED GUIDELINE: Assessment and Control of DNA Reactive (Mutagenic) Impurities In Pharmaceuticals To Limit Potential Carcinogenic Risk M7 (R1).
[7]《化学药品注射剂生产所用的塑料组件系统相容性研究技术指南》(征求意见稿),2020。
